A Brief Introduction to RNA-Seq
RAdelaide 2024
Dr Stevie Pederson
Black Ochre Data Labs
Telethon Kids Institute
Telethon Kids Institute
July 11, 2024
Transcriptomic Analysis
Transcriptomic Analysis
- High-throughput transcriptomic analysis took off ~25 years ago
- Followed the release of the the human genome very rapidly
- Microarrays enabled transcriptome-wide profiling of gene expression
- Probes were designed for each targeted gene
- The Bioconductor package
limmawas first released in 2004 (Smyth 2004)- Still heavily used and maintained
- How does gene expression change across two or more groups
- Control vs Treatment
- Represents an abundance analysis
- Relies on many, many cells pooled together
RNA-Seq
- The development of high-throughput sequencing \(\implies\) RNA-seq
- Rapidly replaced microarrays
- Needed fewer cells \(\implies\) could sample entire transcriptome
- Bulk RNA-Seq built on methods established for microarrays
- New statistical models became quickly established
- Focussed on gene-level analysis
- Now relatively mature
RNA-Seq
- Bulk RNA-Seq at the transcript level is still relatively immature
- Single-Cell RNA-Seq now has a degree of maturity
- Cell trajectory analysis also moving rapidly
- Spatial Transcriptomics is an emerging and rapidly developing technology
- Also includes imaging technologies
- Today we’ll focus on gene-level, bulk RNA-Seq analysis
- Learning file types, packages, object classes, methods
- Also visualisation strategies
Transcriptomes
- Genes are considered as genomic regions transcribed into RNA
- The complete region (i.e. locus) is transcribed
- Introns are spliced out \(\implies\) mature transcripts
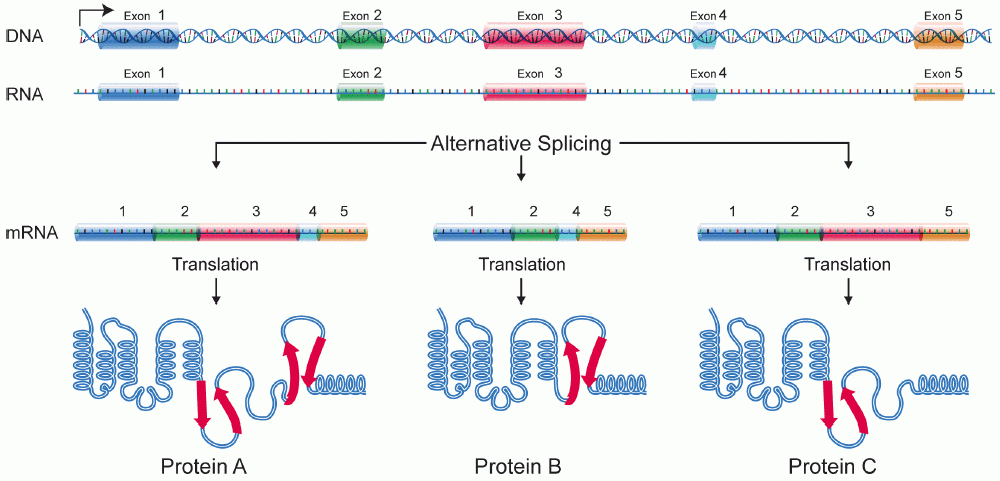
Image courtesy of National Human Genome Research Institute
The Basics of Bulk RNA-Sequencing
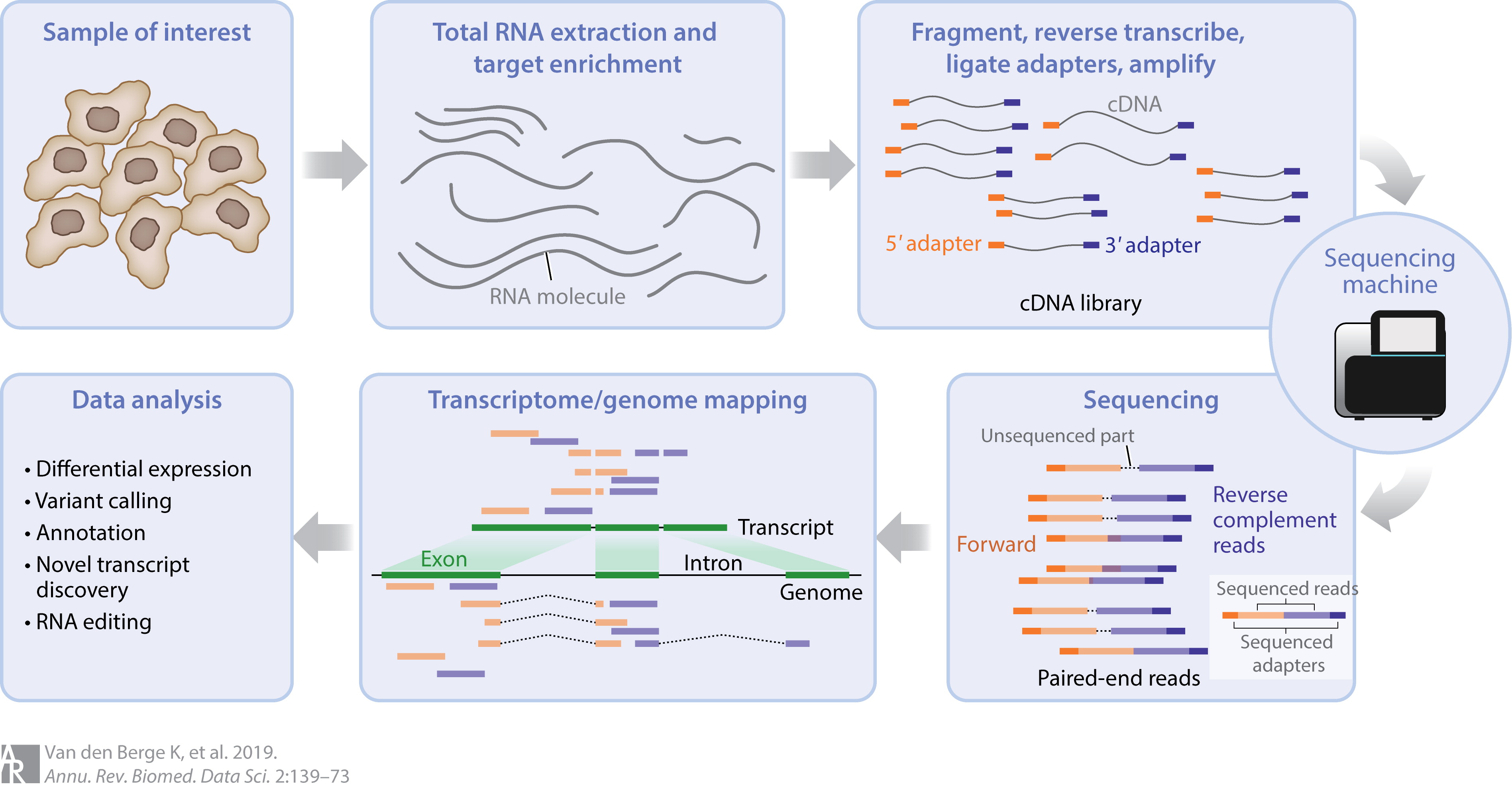
- Intact RNA is extracted from a sample
- Tens of thousands of cells are lysed
- May contain RNA from different cell types
- RNA is fragmented into 250-500bp fragments
- Lose short transcripts \(\implies\) long transcripts broken into pieces
The Basics of Bulk RNA-Sequencing
- Prepared for sequencing
- Converted to cDNA
- Sequencing adapters added to all fragments
- PCR amplification
- Sequenced from both directions (paired end sequencing)
- Commonly 50m reads/sample
The Basics of Bulk RNA-Sequencing
Sequencing Technology
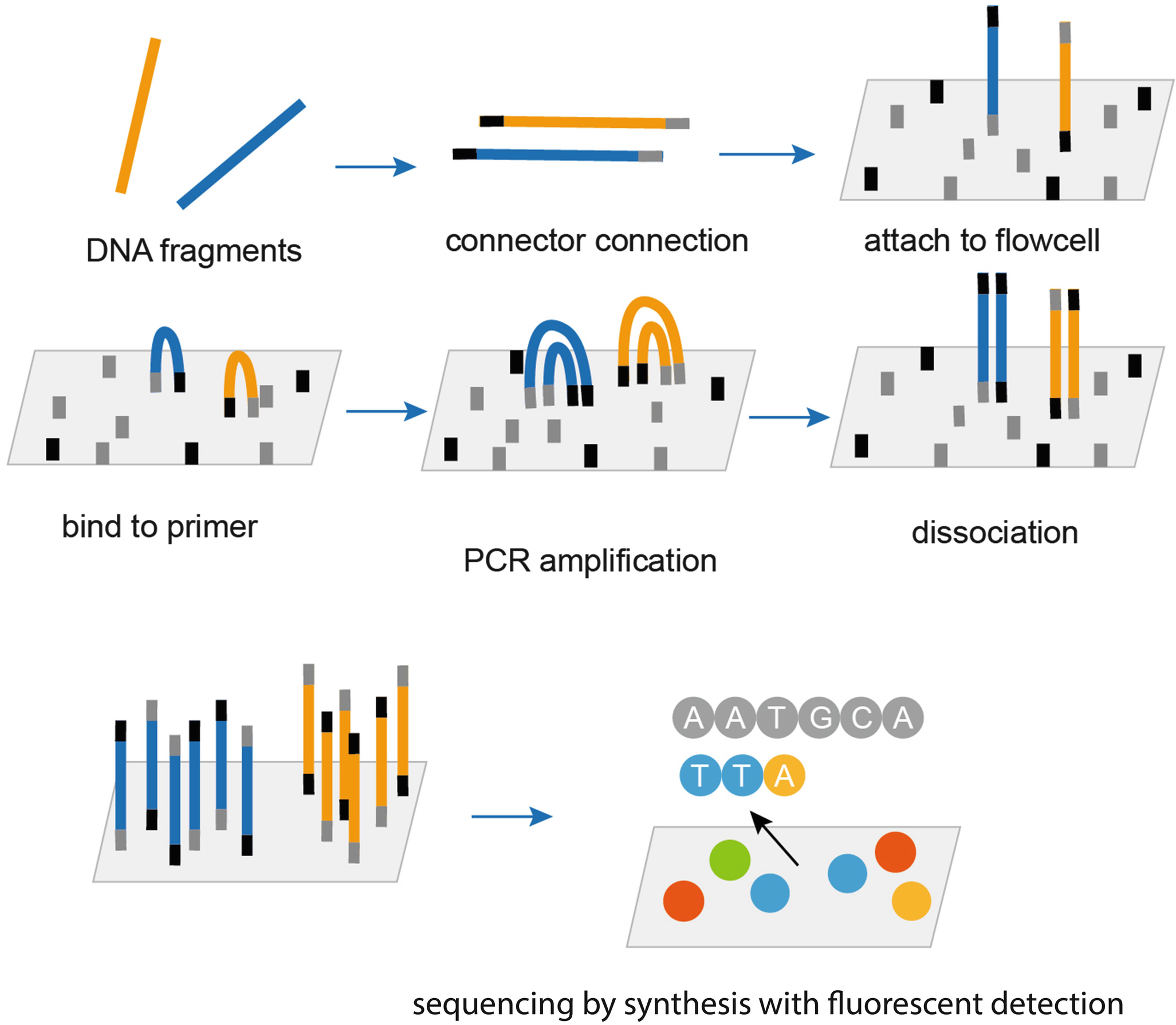
Wang, M. (2021). Next-Generation Sequencing (NGS). In: Pan, S., Tang, J. (eds) Clinical Molecular Diagnostics. Springer, Singapore. https://doi.org/10.1007/978-981-16-1037-0_23
Fastq Files
- The output from sequencing is a
fastqfile- Plain text file
- Commonly 30-50 million reads in each file
- Each read takes 4 lines
- 50m read x 4 lines = 200m lines
- Often compressed with
fq.gzsuffix - Almost never parsed into
R
Fastq Files
- If fragments are sequenced from one end only: single-end reads
- If sequenced from both ends: paired-end reads
- Both provided in separate files
{prefix}_R1.fq.gz+{prefix}_R2.fq.gz- Files match exactly
- Paired reads still represent a single fragment
- More sequence information \(\implies\) more confident alignment
Fastq Files
- Read header starts with
@ - Sequence
+- Quality scores
@SRR001666.1 071112_SLXA-EAS1_s_7:5:1:817:345 length=72
GGGTGATGGCCGCTGCCGATGGCGTCAAATCCCACCAAGTTACCCTTAACAACTTAAGGGTTTTCAAATAGA
+
IIIIIIIIIIIIIIIIIIIIIIIIIIIIII9IG9ICIIIIIIIIIIIIIIIIIIIIDIIIIIII>IIIIII/
@SRR001666.2 071112_SLXA-EAS1_s_7:5:1:801:338 length=72
GTTCAGGGATACGACGTTTGTATTTTAAGAATCTGAAGCAGAAGTCGATGATAATACGCGTCGTTTTATCAT
+
IIIIIIIIIIIIIIIIIIIIIIIIIIIIIIII6IBIIIIIIIIIIIIIIIIIIIIIIIGII>IIIII-I)8IWorkflow Outline
- QC and Read Trimming (optional) 😇
- Align reads to the reference genome 😀
- Count how many reads for each gene 😐
- Statistical (DGE) analysis 😕
- Enrichment analysis 😰
- Biological Interpretation 😱
- Trimming, aligning and counting on an HPC
- QC analysis, Statistical and enrichment analysis in R
- Interpretation with domain experts
Alternative Approaches
- Alternative methods align to transcript sequences NOT the genome
- These alignments are not spliced
- Most reads align to multiple transcripts
- With genomic alignments usually a single alignment
- Transcript-level analysis is a newly solved problem (Baldoni et al. 2024)
- Downstream analysis using transcripts is challenging
- Genes mapped to functions & pathways NOT transcripts
Data Pre-Processing
- A ‘read’ is the total signal from an entire cluster of molecules
- If errors occur during cluster formation:
- Molecules get ‘out-of-sync’
- Actual sequence becomes uncertain
- Low quality scores assigned
- Most people discard low quality reads
- Adapter sequences at either end can also be removed
- Derive from short RNA fragments
Data Pre-Processing
- Overall summaries of library quality can be obtained \(\implies\)
FastQC - Multiple tools to perform Pre-processing
cutadapt,AdapterRemoval,trimmomatic,TrimGaloreetc- Potentially problematic reads or sequences removed
fastpcombines QC reports with read trimming
Data Pre-Processing
FastQC,fastp,cutadaptall return reports after running- Are run on a single library (i.e. sample) at a time
MultiQCis an excellent standalone tool for combining all reportsngsReportsis the “go-to” Bioconductor package for this- Can also import alignment summaries + many others
Alignment
- Pre-processed reads are aligned to a genome
- All aligners based on Burrows-Wheeler Transform
- Comp. Sci algorithm for fast searching
- Requires the creation of an index which is searched
- Most common aligners are
STAR,hisat2orbowtie2- All splice-aware
- GTF used during building of the index
- Requires an HPC & scripting skills
Bam Files
- After alignment to the reference \(\implies\)
bamfiles produced - Usually contain millions of alignments
- Each read now occupies a single line with 11 tab-separated fields
| Column | Field | Description |
|---|---|---|
| 1 | QNAME |
The original FastQ header line |
| 2 | FLAG |
Information regarding pairing, primary alignment, duplicate status, unmapped etc |
| 3 | RNAME |
Reference sequence name (e.g. chr1) |
| 4 | POS |
Left-most co-ordinate in the alignment |
| 5 | MAPQ |
Mapping quality score |
| 6 | CIGAR |
Code summarising exact matches, insertions, deletions etc. |
| 7 | RNEXT |
Reference sequence the mate aligned to |
| 8 | PNEXT |
Left-most co-ordinate the mate aligned to |
| 9 | TLEN |
Read length |
| 10 | SEQ |
The original read sequence |
| 11 | QUAL |
The read quality scores |
Bam Files
- 12th (optional) column often contains tags
NH:i:1indicates this read aligned only onceAS:i:290the actual alignment score produced by the alignerNM:i:2two edits are required to perfectly match the reference
- Reads can align to multiple locations
- Sometimes more alignments than reads
- Rarely, only one read in a pair may align
- Generally view small subsets of data using
samtoolsinbash
Bam Files
- The Bioconductor interface to
bamfiles isRsamtools - We define
BamFileorBamFileListobjects - Are a simple connection to the file (including checks)
- We don’t load anything when created
- Instead define specific subsets to load for a specific task
Bam Files
- When loading a subset of reads set
ScanBamParam() - Enables loading alignments:
- from specific ranges (using
GRanges) - with specific tags or flags
- from specific ranges (using
- Controls which columns are returned
- Sequences loaded as
DNAStringSets - Alignment co-ordinates as
GRangesobjects
Read Counting
- Most assigning of reads to genes is performed by external tools
- Gene, Transcript & Exon co-ordinates passed in a GTF
- Aligned Reads overlapping an exon/transcript/gene are counted
- Produces flat (i.e. tsv) files \(\implies\) count once & load each time you start
- Today’s count data produced using
featureCountsfrom theSubreadtool- Other tools are
RSEM,htseq
- Other tools are
Read Counting
- Counting can be performed in
R:RsubreadorGenomicAlignments
- Should still only be performed once and saved as a standalone file
- Would avoid doing at the start of every R script
Reference Annotations
- Reference genomes are relatively stable over many years
- Available from multiple sources
- Inconsistent use of the “chr” prefix
- Inconsistency in “chrM” or “chrMT”
- Some variability in scaffolds
Reference Annotations
- Annotation sets are updated multiple times every year
- Gencode, Ensembl, UCSC
- Usually minor changes
- Not important for genomic alignment
- Very important for read counting & transcriptomic alignment
Reference Annotations
- The latest Gencode set of annotations (Release 46, May 2024)
- 63,086 Genes
- 19,411 protein coding
- Remainder are lncRNA, pseudogenes etc
- 254,070 Transcripts
- 89,581 protein coding
- Remainder are NMD, lncRNA etc
- Shortest annotated (spliced) transcript is 8nt \(\implies\) longest is 350,375nt
References
![]()
Baldoni, Pedro L, Yunshun Chen, Soroor Hediyeh-Zadeh, Yang Liao, Xueyi Dong, Matthew E Ritchie, Wei Shi, and Gordon K Smyth. 2024. “Dividing Out Quantification Uncertainty Allows Efficient Assessment of Differential Transcript Expression with edgeR.” Nucleic Acids Res. 52 (3): e13.
Smyth, Gordon K. 2004. “Linear Models and Empirical Bayes Methods for Assessing Differential Expression in Microarray Experiments.” Stat. Appl. Genet. Mol. Biol. 3 (February): Article3.